Comparative Transcriptomics Provides Insight into Floral Color Polymorphism in a Pleione limprichtii Orchid Population

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Color Differences and Quantity Distribution Pattern
2.2. Major Anthocyanin Compounds in P. limprichtii
2.3. RNA-Seq and Annotation of Unigenes
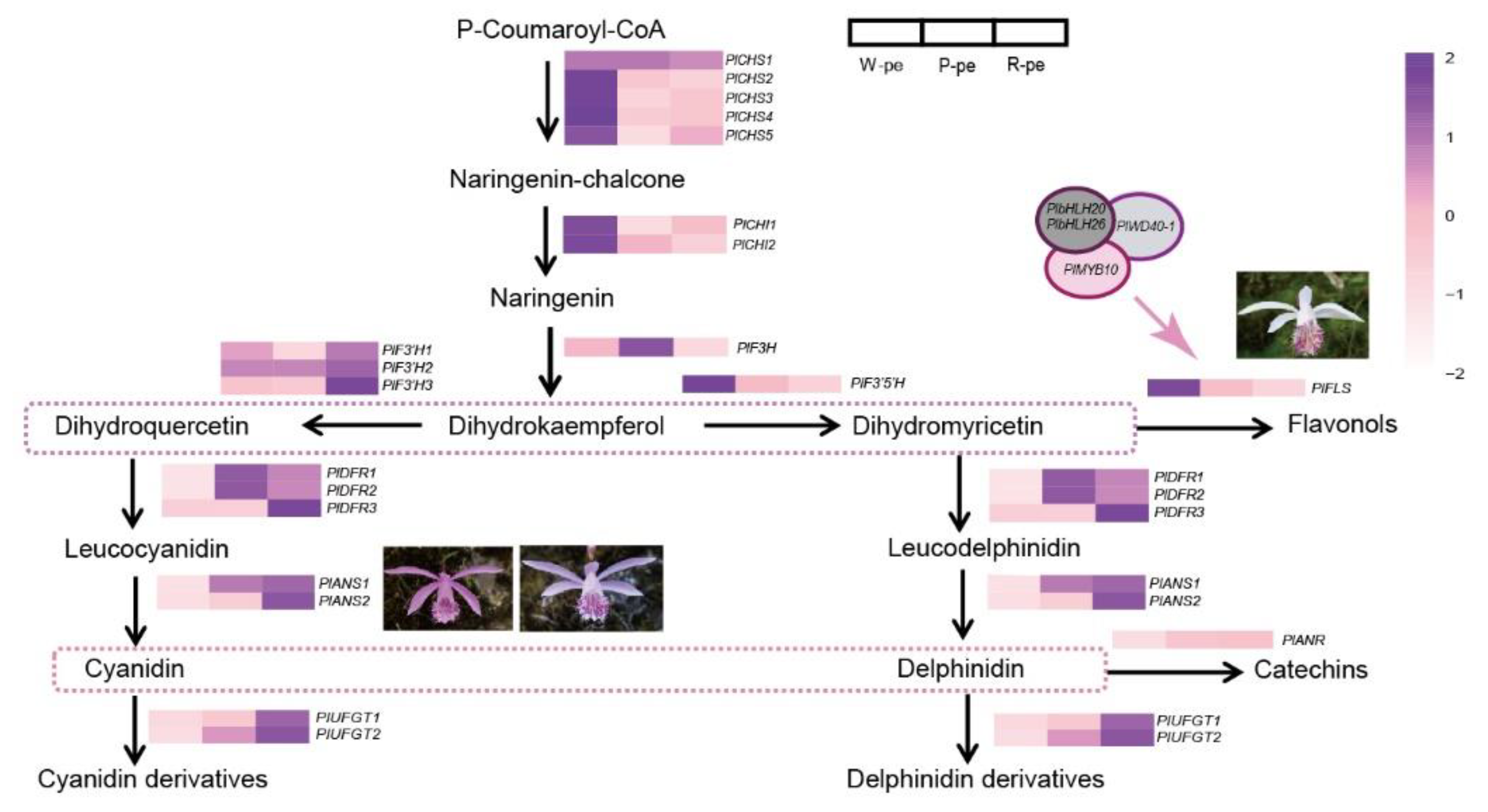
2.4. The Expression Patterns of ABP Genes
2.5. The Relationship between Structure Genes and TFs
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Quantitative Statistics and Flower Colorimeter Analysis
4.3. Sampling and RNA Extraction and cDNA Synthesis
4.4. Measurement of Flower Anthocyanin
4.5. Library Preparation and Sequencing
4.6. De Novo Transcriptome Assembly Annotation
4.7. Expression Profile and RT-qPCR
4.8. Genes Related to the ABP and Phylogenetic Analyses
4.9. Structure Genes and Tfs Co-Expression Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| UPLC | Ultra performance liquid chromatography |
| ABP | Anthocyanin biosynthesis pathway |
| CHS | Chalcone synthase |
| CHI | Chalcone isomerase |
| F3H | Flavanone 3-hydroxylase |
| F3′H | Flavonoid 3’-hydroxylase |
| F3′5′H | Flavonoid 3’5′-hydroxyla |
| FLS | Flavonol synthase |
| DFR | Dihydroflavonol 4-reduct |
| ANS | Anthocyanidin synthase |
| ANR | Anthocyanidin reductase |
| UFGT | UDP flavonoid glucosyl transferase |
| RHS | Royal horticulture society |
| RHSCC | The royal horticulture society color chart |
| NR | Non-redundant protein database |
| Swiss-Prot | Swiss-Protein protein database |
| KEGG | Kyoto encyclopedia of genes and genomes |
| KOG | Eukaryotic orthologous groups |
| GO | Gene ontology |
| RPKM | reads per kb per million reads |
| FDR | false discovery rate |
| qRT-PCR | Real-time reverse transcription-PCR |
References
- Cooley, A.M.; Carvallo, G.; Willis, J.H. Is floral diversification associated with pollinator divergence? flower shape, flower colour and pollinator preference in Chilean Mimulus. Ann. Bot. 2008, 101, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.M.; Albert, N.W.; Schwinn, K.E. From landing lights to mimicry: The molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct. Plant Biol. 2012, 39, 619–638. [Google Scholar] [CrossRef]
- Vaidya, P.; Ansley, M.; Emily, M.; Michaela, K.; Lauren, C.; Lee, C.R.; Bingham, R.A.; Anderson, J.T. Ecological causes and consequences of flower color polymorphism in a self-pollinating plant (Boechera Stricta). New Phytol. 2018, 218, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streinzer, M.; Roth, N.; Paulus, H.F.; Spaethe, J. Color preference and spatial distribution of glaphyrid beetles suggest a key role in the maintenance of the color polymorphism in the peacock anemone (Anemone pavonina, Ranunculaceae) in Northern Greece. J. Comp. Physiol. 2019, 205, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, R.T.; Byers, K.J.R.P.; De Brito Francisco, R.M.; Staedler, Y.M.; LaFountain, A.M.; Schönenberger, J.; Schiestl, F.P.; Schlüter, P.M. Emergence of a floral colour polymorphism by pollinator-mediated overdominance. Nat. Commun. 2019, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel, J.M.; Matthew, A.S. Flower color as a model system for studies of plant evo-devo. Front. Plant Sci. 2013, 4, 321. [Google Scholar] [CrossRef] [PubMed]
- Rausher, M.D. Evolutionary transitions in floral color. Int. J. Plant Sci. 2008, 169, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Lou, Q.; Liu, Y.L.; Qi, Y.Y.; Jiao, S.Z.; Tian, F.F.; Jiang, L.; Wang, Y.J. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower color in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar] [CrossRef] [Green Version]
- Butler, T.; Dick, C.; Carlson, M.L.; Whittall, J.B. Transcriptome analysis of a petal anthocyanin polymorphism in the Arctic Mustard, Parrya Nudicaulis. PLoS ONE 2014, 9, e101338. [Google Scholar] [CrossRef] [Green Version]
- Casimiro-Soriguer, I.; Eduardo, N.; Buide, M.L.; Valle, J.C.; Whittall, J.B. Transcriptome and biochemical analysis of a flower color polymorphism in Silene Littorea (Caryophyllaceae). Front. Plant Sci. 2016, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.X.; Yang, H.X.; Liu, H.F.; Yang, J.; Hu, Y.H. Extensive transcriptome changes underlying the flower color intensity variation in Paeonia Ostii. Front. Plant Sci. 2016, 6, 1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lucie, C.C.; Bessière, J.M.; Guillaume, C.; Bertrand, S.; Eric, I. Flower color polymorphism in Iris Lutescens (Iridaceae): Biochemical analyses in light of plant–insect interactions. Phytochemistry 2013, 94, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.A.; Matthew, A.S.; Laura, I.N.; Kaitlyn, A.C. The genetic basis of a rare flower color polymorphism in Mimulus Lewisii provides insight into the repeatability of evolution. PLoS ONE 2013, 8, e81173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotewold, E. The genetic and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Holton, T.A.; Edwina, C.C. Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell 1995, 7, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Tao, J. Recent advances on the development and regulation of flower color in ornamental plants. Front. Plant Sci. 2015, 6, 261. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Akemi, O. Seeing is believing: Engineering anthocyanin and carotenoid biosynthetic pathways. Curr. Opin. Biotechnol. 2008, 19, 190–197. [Google Scholar] [CrossRef]
- Xie, D.Y.; Shashi, B.S.; Elane, W.; Wang, Z.Y.; Richard, A.D. Metabolic engineering of proanthocyanidins through co-expression of anthocyanidin reductase and the PAP1 MYB transcription factor. Plant J. 2006, 45, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Twyford, A.D.; Aaron, M.C.; Pratibha, C.; Ramesh, R.; Jannice, F. Loss of color pigmentation is maintained at high frequency in a monkey flower population. Am. Nat. 2018, 191, 135–145. [Google Scholar] [CrossRef]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [Green Version]
- Albert, N.W.; Kevin, M.D.; David, H.L.; Zhang, H.B.; Mirco, M.; Cyril, B.; Boase, M.R.; Hanh, N.; Jameson, P.E.; Schwinn, K.E. A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in Eudicots. Plant Cell 2014, 26, 962–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessinger, C.A.; Rausher, M.D. Lessons from flower color evolution on targets of selection. J. Exp. Bot. 2012, 63, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cribb, P.J.; Gale, S.W. “Pleione”. In the Flora of China 25 (Orchidaceae); Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Beijing Science Press and Missouri Botanical Garden Press: Beijing, China, 2009; pp. 325–333. [Google Scholar]
- Cribb, P.; Butterfield, I. The genus Pleione, 2nd ed.; Royal Botanic Gardens/Kew Press: London, UK, 1999; p. 27. [Google Scholar]
- Govaerts, R.; Campacci, M.A.; Baptista, D.H.; Cribb, J.; George, A.; Kreutz, K. Data from: World Checklist of Orchidaceae; The Board of Trustees of the Royal Botanic Gardens/Kew: London, UK, 2016; Available online: http://apps.kew.org/wcsp (accessed on 20 August 2019).
- Gravendeel, B.; Eurlings, M.C.M.; Berg, C.V.D.; Cribb, P.J. Phylogeny of Pleione (Orchidaceae) and parentage analysis of its wild hybrids based on plastid and nuclear ribosomal its sequences and morphological data. Syst. Bot. 2004, 29, 50–63. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis Thaliana. Curr. Opin. Biotechnol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Dubos, C.; Ralf, S.; Grotewold, E.; Weisshaar, B.; Martin, C.; Loïc, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Arista, M.; Talavera, M.; Berjano, R.; Ortiz, P.L. Abiotic factors may explain the geographical distribution of flower colour morphs and the maintenance of colour polymorphism in the scarlet pimpernel. J. Ecol. 2013, 101, 1613–1622. [Google Scholar] [CrossRef]
- Imbert, E.; Wang, H.; Conchou, L.; Vincent, H.; Talavera, M.; Schatz, B. Positive effect of the yellow morph on female reproductive success in the flower colour polymorphic Iris lutescens (Iridaceae), a deceptive species. J. Evol. Biol. 2014, 27, 1965–1974. [Google Scholar] [CrossRef]
- Chalker-Scott, L. Environmental significance of anthocyanins in plant stress responses. Photochem. Photobiol. 1999, 70, 1–9. [Google Scholar] [CrossRef]
- Muchhala, N.; Sönke, J.; Smith, S.D. Competition for hummingbird pollination shapes flower color variation in Andean solanaceae: Competition for pollination shapes flower color variation. Evolution 2014, 68, 2275–2286. [Google Scholar] [CrossRef]
- Parachnowitsch, A.L.; Christina, M.C. Predispersal seed herbivores, not pollinators, exert selection on floral traits via female fitness. Ecology 2008, 89, 1802–1810. [Google Scholar] [CrossRef]
- Jin, X.H.; Huang, H.; Wang, L.; Sun, Y.; Dai, S.L. Transcriptomics and metabolite analysis reveals the molecular mechanism of anthocyanin biosynthesis branch pathway in different senecio cruentus cultivars. Front. Plant Sci. 2016, 7, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Maitre, N.C.; Pirie, M.D.; Bellstedt, D.U. Floral color, anthocyanin synthesis gene expression and control in cape erica species. Front. Plant Sci. 2019, 10, 1565. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhai, Y.; Luo, X.; Zhang, Y.; Shi, Q.Q. Comparative transcriptome analyses reveal genes related to pigmentation in the petals of red and white Primula vulgaris cultivars. Physiol. Mol. Biol. Plants 2019, 25, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.W.; Alexandra, B.R.; Janelle, M.S.; Lauren, E.S.; Harvey, D.B. Competition between anthocyanin and flavonol biosynthesis produces spatial pattern variation of floral pigments between Mimulus species. Proc. Natl. Acad. Sci. USA 2016, 113, 2448–2453. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.M.; Schwinn, K.E.; Deroles, S.C.; Manson, D.G.; Lewis, D.H.; Bloor, S.J.; Bradley, J.M. Enhancing anthocyanin production by altering competition for substrate between flavonol synthase and dihydroflavonol 4-Reductase. Euphytica 2003, 131, 259–268. [Google Scholar] [CrossRef]
- Sangkyu, P.; Kim, D.H.; Lee, J.Y.; Ha, S.H.; Lim, S.H. Comparative analysis of two flavonol synthases from different-colored onions provides insight into flavonoid biosynthesis. J. Agric. Food Chem. 2017, 65, 5287–5298. [Google Scholar]
- Shi, S.G.; Li, S.J.; Kang, Y.X.; Liu, J.J. Molecular characterization and expression analyses of an anthocyanin synthase gene from magnolia sprengeri Pamp. Int. J. Appl. Biotechnol. Biochem. 2015, 175, 477–488. [Google Scholar] [CrossRef]
- Wu, X.X.; Gong, Q.H.; Ni, X.P.; Zhou, Y.; Gao, Z.H. UFGT: The key enzyme associated with the petals variegation in Japanese Apricot. Front. Plant Sci. 2017, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Feller, A.; Machemer, K.; Edward, L.B.; Grotewold, E. Evolutionary and comparative analysis of MYB and BHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef]
- Heim, M.A. The basic Helix-Loop-Helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Baudry, A.; Heim, M.A.; Dubreucq, B.; Caboche, M.; Weisshaar, B.; Lepiniec, L. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis Thaliana. Plant J. 2004, 39, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, I.; Heim, M.; Weisshaar, B.; Uhrig, J. Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like bHLH proteins: Systematic analysis of MYB/BHLH-Interaction. Plant J. 2004, 40, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Ikegami, A.; Tsujimoto, T.; Kobayashi, S.; Sato, A.; Kono, A.; Yonemori, K. DkMyb4 is a myb transcription factor involved in proanthocyanidin biosynthesis in persimmon fruit. Plant Physiol. 2009, 151, 2028–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, K.; Yoshimi, U.; Masaru, O.T. AtMYBL2, a protein with a single MYB domain, acts as a negative regulator of anthocyanin biosynthesis in Arabidops. Plant J. 2008, 55, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.A.; Li, N.; Li, L.F.; Zhang, S.Z. Integrated metabolome and transcriptome analysis uncovers the role of anthocyanin metabolism in Michelia maudiae. Int. J. Genom. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Zhu, J.; Hao, Q.; Yuan, Y.; Duan, Y.; Men, S.; Wang, Q.; Hou, Q.; Liu, Z.; Shu, Q.; et al. A novel R2R3-MYB transcription factor contributes to petal blotch formation by regulating organ-Sspecific expression of PsCHS in tree peony (Paeonia Suffruticosa). Plant Cell Physiol. 2018, 60, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gong, L.; Guo, Z.L.; Wang, W.S.; Zhang, H.Y.; Liu, X.Q.; Yu, S.B.; Xiong, L.Z.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Rose-Purple | Pink | White |
|---|---|---|---|
| population1 | 409 (58%) | 227 (32%) | 70 (10%) |
| population2 | 31 (65%) | 15 (31%) | 2 (4%) |
| population3 | 92 (60%) | 54 (35%) | 8 (5%) |
| Flower Color | RHS | L* | C* | b*/a* |
|---|---|---|---|---|
| purple red | Purple-group N78B | 67.92 ± 10.27 | 49.64 ± 9.41 | −1.68 ± 0.61 |
| 76.66 ± 4.51 | 52.88 ± 4.18 | −2.35 ± 0.23 | ||
| 67.35 ± 10.34 | 57.13 ± 6.24 | −1.68 ± 0.61 | ||
| 81.31 ± 8.72 | 51.89 ± 7.42 | −1.91 ± 0.44 | ||
| 79.68 ± 15.55 | 46.28 ± 11.76 | −1.85 ± 0.79 | ||
| Pink | Purple-group 75A | 61.08 ± 5.29 | 26.71 ± 1.82 | −1.20 ± 0.36 |
| 66.05 ± 6.63 | 35.45 ± 9.60 | −1.77 ± 0.71 | ||
| 66.35 ± 2.73 | 31.72 ± 3.14 | −1.57 ± 0.29 | ||
| 58.80 ± 5.20 | 29.66 ± 6.05 | −2.20 ± 1.07 | ||
| 62.80 ± 5.78 | 25.43 ± 4.99 | −2.56 ± 0.89 | ||
| White | Purple-group 76D | 93.08 ± 5.57 | 13.31 ± 9.67 | −4.04 ± 2.06 |
| 88.12 ± 8.13 | 18.53 ± 7.31 | −13.60 ± 5.70 | ||
| 95.85 ± 4.48 | 22.41 ± 6.77 | −7.97 ± 3.88 | ||
| 87.52 ± 11.45 | 25.27 ± 10.21 | −6.11 ± 2.61 | ||
| 87.07 ± 7.97 | 21.36 ± 6.12 | −9.28 ± 4.73 |
| Index | Ion Mode | Molecular Weight | Substance |
|---|---|---|---|
| pme3609 | positive | 287.24 | Cyanidin |
| pme0442 | positive | 303.24 | Delphinidin |
| pmb0541 | positive | 697.1 | Cyanidin 3-O-glucosyl-malonylglucoside |
| pmb0542 | positive | 535.1 | Cyanidin 3-O-malonylhexoside |
| Pathway | Gene | Encoding Enzyme | Number |
|---|---|---|---|
| Flavonoid biosynthesis | CHS | Chalcone synthase | 5 |
| CHI | Chalcone isomerase | 2 | |
| F3H | Flavanone 3-dioxygenase | 1 | |
| F3′H | Flavonoid 3’-hydroxylase | 3 | |
| F3′5′H | Flavonoid 3’,5’-hydroxylase | 1 | |
| DFR | Dihydroflavonol 4-reductase | 3 | |
| ANS | Anthocyanidin synthase | 2 | |
| ANR | Anthocyanidin reductase | 1 | |
| FLS | Flavonol synthase | 1 | |
| Anthocyanin biosynthesis | UFGT | Anthocyanidin 3-O-glucosyltransferase | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhou, T.; Dai, Z.; Dai, X.; Li, W.; Cao, M.; Li, C.; Tsai, W.-C.; Wu, X.; Zhai, J.; et al. Comparative Transcriptomics Provides Insight into Floral Color Polymorphism in a Pleione limprichtii Orchid Population. Int. J. Mol. Sci. 2020, 21, 247. https://doi.org/10.3390/ijms21010247
Zhang Y, Zhou T, Dai Z, Dai X, Li W, Cao M, Li C, Tsai W-C, Wu X, Zhai J, et al. Comparative Transcriptomics Provides Insight into Floral Color Polymorphism in a Pleione limprichtii Orchid Population. International Journal of Molecular Sciences. 2020; 21(1):247. https://doi.org/10.3390/ijms21010247
Chicago/Turabian StyleZhang, Yiyi, Tinghong Zhou, Zhongwu Dai, Xiaoyu Dai, Wei Li, Mengxia Cao, Chengru Li, Wen-Chieh Tsai, Xiaoqian Wu, Junwen Zhai, and et al. 2020. "Comparative Transcriptomics Provides Insight into Floral Color Polymorphism in a Pleione limprichtii Orchid Population" International Journal of Molecular Sciences 21, no. 1: 247. https://doi.org/10.3390/ijms21010247